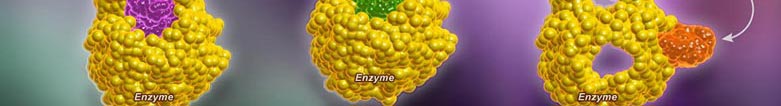



Enzyme inhibitors prevent the formation of an enzyme-substrate complex and hence prevent the formation of product. Inhibition of enzymes may be either reversible or irreversible depending on the specific effect of the inhibitor being used.
In a normal reaction, a substrate binds to an enzyme (via the active site) to form an enzyme-substrate complex. The shape and properties of the substrate and active site are complementary, resulting in enzyme-substrate specificity. When binding occurs, the active site undergoes a conformational change to optimally interact with the substrate. This conformational change destabilizes chemical bonds within the substrate, lowering the activation energy. As a consequence ofthis enzyme interaction, the substrate is converted into product at an accelerated rate.
A competitive inhibition involves a molecule, other than the substrate, binding to the enzyme’s active site. The molecule (inhibitor) is structurally and chemically similar to the substrate (hence able to bind to the active site). The competitive inhibitor blocks the active site and thus prevents substrate binding. As the inhibitor is in competition with the substrate, its effects can be reduced by increasing substrate concentration.
A non-competitive inhibition involves a molecule binding to a site other than the active site (an allosteric site). The binding of the inhibitor to the allosteric site causes a conformational change to the enzyme’s active site. As a result of this change, the active site and substrate no longer share specificity, meaning the substrate cannot bind. As the inhibitor is not in direct competition with the substrate, increasing substrate levels cannot mitigate the inhibitor’s effect.
Drugs that function as enzyme inhibitors constitute a significant portion of the orally bioavailable therapeutic agents that are in clinical use today. Most of the drug discovery and development efforts at present are focused on identifying and optimizing drug candidates that act through inhibition of specific enzyme targets. The attractiveness of enzymes as targets for drug discovery stems from the high levels of disease association (target validation) and druggability (target tractability) that typically characterize this class of proteins.
Aurora Medbiochem is proud to offer a variety of structurally diverse enzyme inhibitors for research.
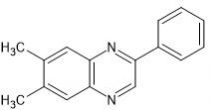
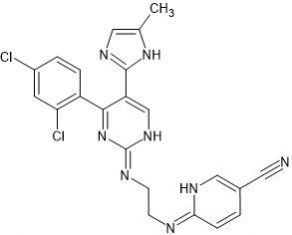
AG-1295 is a selective platelet-derived growth factor receptor (PDGFR) tyrosine-kinase inhibitor.
A potent thiadiazolyl analog that binds to PH (pleckstrin homology) domain (Kd = 40.8 µM, Ki = 2.4 µM)..
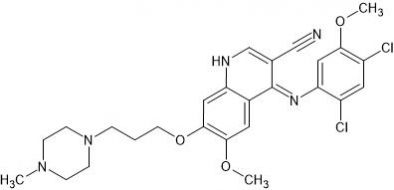
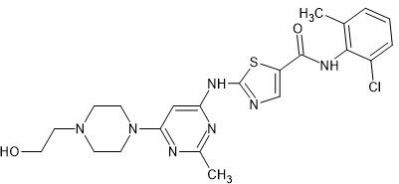
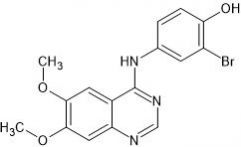
Bosutinib (SKI-606) is a novel tyrosine kinase inhibitor. Bosutinib can overcome not only Bcr-Abl-dependent mechanisms of resistance, but also those that are Bcr-Abl-independent. It is used to treat patients with chronic myelogenous leukemia (CML) who demonstrate resistance to Imatinib or develop resistance during treatment.
CVT-313 is a potent, cell-permeable, selective, reversible, and ATP-competitive inhibitor of Cdk2 (IC50 = 0.5 µM for Cdk2/A and Cdk2/E; 4.2 µM for Cdk1/B; 215 µM for Cdk4/D1). Inhibits other kinases only at much higher concentrations (IC50 >1.25 mM for MAPK, PKA, and PKC).
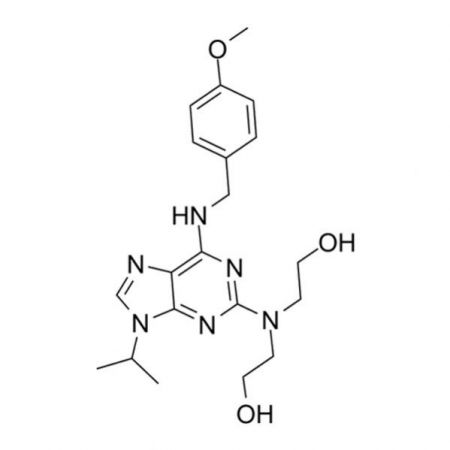
A potent and highly selective inhibitor of glycogen synthase kinase-3β (GSK-3β) (IC₅₀ = 6.7 nM). CHIR99021 has been shown to allow for long-term expansion of murine embryonic stem cells in a chemically-defined medium in conjunction with MEK/MAPK inhibitor PD184352 and fibroblast growth factor receptor (FGFR) inhibitor SU5402.
A 10 mM (4.65 mg in 1 ml), sterile-filtered solution of the GSK-3β inhibitor CHIR99021 (Cat. No. AUR10277) in anhydrous DMSO.
DAPK inhibitor is a potent, ATP-competitive, and highly selective death-associated protein kinase (DAPK) inhibitor (IC50 = 69 and 225 nM against DAPK1 and DAPK3, respectively, with 10 µM ATP), while exhibiting much reduced or no activity against a panel of 48 other kinases even at concentrations as high as 10 µM.
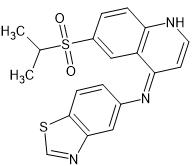
Dasatinib is a multi-kinase inhibitor with potent activity against BCR–ABL kinase (IC₅₀ <1 nM) and Src family kinases (IC₅₀ = 0.2–1.1 nM). Dasatinib also inhibits Lyn (IC₅₀ = 8.5 nM) and Src (IC₅₀ = 3.0 nM) kinase activities in vitro using 0.1 mg/mL poly (Glu4-Tyr) as the substrate.
GSK-872 is an inhibitor of receptor-interacting protein kinase 3 (RIPK3). It is selective for RIPK3, having >1,000-fold selectivity over 300 kinases in a fluorescence polarization assay at a concentration of 1 μM. GSK872 inhibits TNF- and virus-induced necrosis in 3T3-SA fibroblasts.
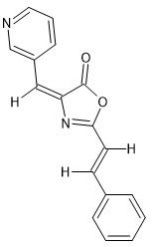
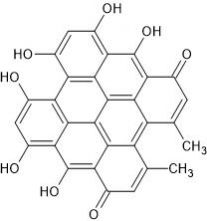
Hypericin is an active ingredient in the medical herb St. John’s Wort (SJW). Hypericin displays antineoplastic activity as well as antibiotic and antiviral activities.
WHI-P154 is a potent, cell-permeable, reversible, ATP-competitive, and specific inhibitor of JAK3 (IC50 = 5.6 µM). Has no effect on either JAK1 or JAK2.
LIMKi 3 is a cell-permeable compound that acts as a potent LIM kinase inhibitor (IC50 = 7 and 8 nM against LIMK1 and LIMK2, respectively) and effectively suppresses cellular cofilin phosphorylation (IC50 ~ 1 µM in A549 and MDA-MB-231 cultures) without affecting tubulin polymerization or inducing cytotoxicity (EC50 >10 µM in A549 proliferation and colony formation assays).